

github download link: (simrlls)
This post has been modified for the updated v.0.0.8 script.
The program simrrls can be used to simulate raw (fastq format) sequence data on an input topology under the coalescent in a manner that emulates restriction-site associated DNA, with slight variations for different data types (e.g., RADseq, ddRAD, GBS, paired-end data).
It was originally developed to generate data for bug testing pyrad and to create example data sets for use in tutorials. However, I can imagine other uses for it, such as investigating the effects of missing data, insufficient sequencing coverage and error rates on assembly results.
To use the program you will need to first install the Egglib Python module, used here to perform coalescent simulations.
Calling the script – simrrls
$ simrrls -h
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-o outname [str] output file name prefix (default 'out')
-mc dropout [0/1] allelic dropout from mutation to cut sites (default 0)
-ms dropout [0/1] allelic dropout from new cut sites in seq (default 0)
-e error [float] sequencing error rate (default 0.0005)
-f datatype [str] datatype (default rad) (options: rad, gbs, ddrad,
pairddrad, pairgbs)
-I indels [float] rate of indel mutations (default 0) ex: 0.001
-l length [int] length of simulated sequences (default 100)
-L nLoci [int] number of loci to simulate (default 100)
-n Ninds [int] N individuals from each taxon (default 1)
-N Ne [int] pop size (Ne for all lineages; default 5e5)
-t tree [str] file name or newick string of ultrametric tree
(default 12 taxon balanced tree w/ bls=1)
-u mu [float] per site mutation rate (default 1e-9)
-df depthfunc [str] model for sampling copies (default norm, other=exp)
-dm depthmean [int] mean sampled copies in norm, 1/m for exp (default 10)
-ds depthstd [int] stdev sampled copies, used with norm model (default 0)
-c1 cut_1 [str] restriction site 1 (default CTGCAG)
-c2 cut_2 [str] restriction site 1 (default CCGG)
-i1 min_insert [int] total frag len = (2*l)+insert (default 100)
-i2 max_insert [int] total frag len = (2*l)+insert (default 400)
-r1 seed_1 [int] random seed 1 (default 1234567)
-r2 seed_2 [int] random seed 2 (default 7654321)Datatypes– RAD is the default type for which a barcode and restriction site overhang are attached to the left side of single end reads. Depending on the data type selected simrrls will put cut sites and barcodes in the proper ends of sequences so that they emulate RAD, GBS, or ddRAD data, single or paired-end. The choice also effects how locus dropout occurs (i.e., one or two cutters present).
Allelic Dropout– There are two ways in which allelic dropout (mutation-disruption) can occur, each of which can be toggled. The first is mutations to the cut site recognition sequence (-mc). If this is turned on then an allele will be dropped if a mutation occurs within the length of the restriction recognition sites determined by the cutters (-c1 and -c2) and the data type (-f) which determines whether one or two cutters are used (e.g., rad is a single cutter, ddrad is double-cutter). The other form of mutation disruption occurs when mutations give rise to new cut sites within the length of a selected fragment (-ms). Here the total fragment length is determined by (-l) and the max insert length (-i2), and the probability of disruption by the cutters (-c1 and -c2), and the substitution rate (-N and -u).
Sequencing coverage and error– Another source of missing data in RADseq data sets is low sequencing coverage. You can set the sampling rate as the number of reads that are sampled from each haplotype. The mean (-dm) and standard deviation (-ds). To examine the effect sequencing rate has on base calls, etc., you could combine low coverage sequencing with a high error rate (-e).
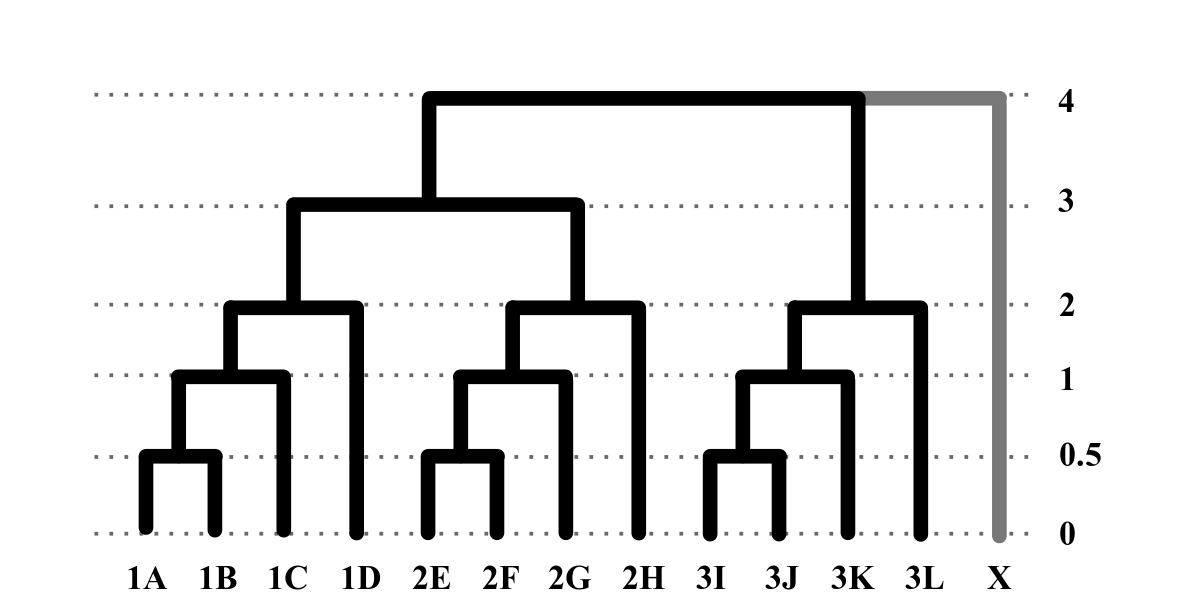
Topology– Data are simulated under a Jukes-Cantor model with equal base frequencies. If there is no user-supplied topology (-t) then simrrls uses a default 12 tip topology, shown below. Branch lengths are in coalescent units. The outgroup taxon in the tree “X” is not included in the output, but is used to polarize mutations relative to an outgroup for creating indels.
Output – Two data files are created using the output name
prefix (-o), which if it were ‘testrad’ would create
testrad_R1_.fastq.gz and testrad_barcodes.txt. The first is a
compressed fastq file with sequence data and the
latter is a text file mapping barcodes to sample names.
If you selected paired-end data it would create
two sequence files, one with “_R1_”
in the name and the other with “_R2_”.
With these fastq data and a barcode map the data can then be
assembled in pyrad.
Examples –
Modified population parameters:
$ simrrls -o test2 -N 1e6 -u 2e-8Modified sequencing parameters:
$ simrrls -o test3 -L 5000 -l 200 -e 0.001 -dm 10 -ds 2Modified library type (In this case allowing paired-end reads overlap):
$ simrrls -o test4 -f pairddrad -i1 -50 -i2 200Modified topology:
$ echo "((a:1,b:1):1,c:2);" > treefile
$ simrrls -o test5 -t treefileMore info – Check out the git repository: (simrlls.py)